
二代测序使寻找基因突变变得更容易了,科研者可以很方便地获得任何个体的整个基因组范围内的突变。但是医学问题并没有因此得到解决,因为存在大量无关紧要的突变,这是基因多样性的一种表现,而非病理性的异常。此外对于肿瘤疾病而言,基因组上存在大量的突变,筛选关键突变成为重中之重,无论对于病因学还是基因靶向治疗而言都是必需的。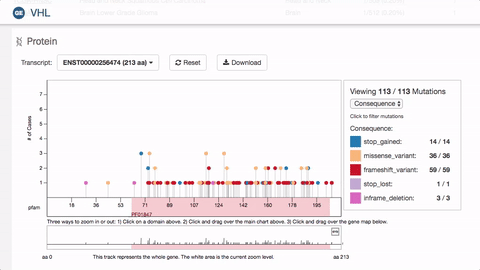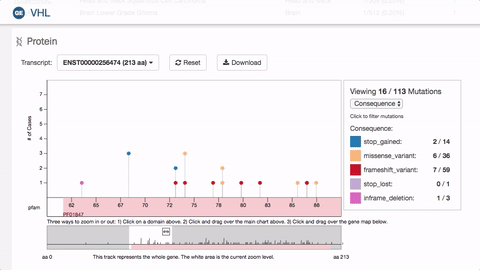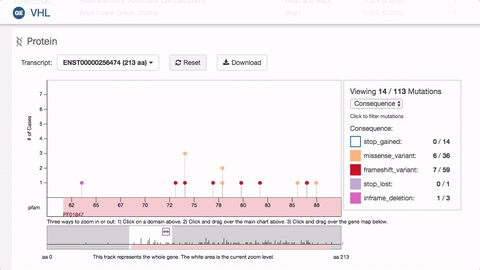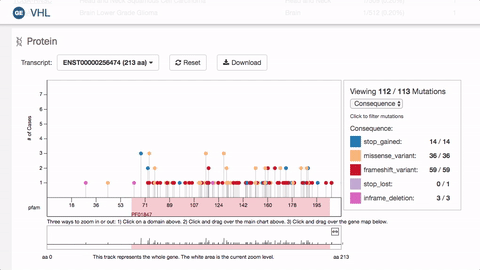
基因突变的寻找和功能评估的流程如下:
Step 1: 寻找突变
寻找突变的工具Mutation Calling工具:
GATK4,这是最为常用的
Varscan
SomaticSniper
Strelka
Step 2: 突变的重要性
- 基于疾病数据库进行注释: 看该突变是否被数据库收录,如OMIM、COSMIC等
- 基于数据本身统计特征: 依赖于群体的数据,多数依赖于肿瘤-正常组织配对设计
- MutSig/MutSigCV: 统计突变频率高于基因组背景突变频率的基因,以此发现驱动基因。基于MATLAB的工具
- MSEA: 通过分析突变热点区域(Mutation hotspot region)发现候选驱动基因
- 基于DNA序列的进化保守性:
- SIFT: 根据序列的保守性预测突变对蛋白质功能的影响。保守性越大,该位点功能越重要,突变的危害越大。而保守性数据基于PSI-BLAST对同源序列比对的结果。
- 基于DNA序列的基因组注释:
- ANNOVAR: 能够确定突变是否导致蛋白质编码变化以及受影响的氨基酸,也能识别特定基因组区域的variants,如保守性区域,TFBS,DHS,组蛋白结合修饰区域。
- Oncotator: ANNOVAR的竞争者
- SnpEff:
- VEP:
- 基于蛋白质的结构和功能: (同时也考虑了DNA序列的特征)
- PolyPhen: 除了序列本身的保守性外,还参考突变对蛋白质三维结构和功能的影响。
- STRUM: 预测突变对蛋白质折叠稳定性的影响
- CanDra: 基于蛋白质结构、进化和基因特征。参考了COSMIC数据库
- MutationTaster: 基于蛋白质结构、突变率、OMIM数据库、HGMD数据库
- ActiveDriver:基于蛋白质的活跃区,如信号位点、结构域、调控区域等
- CanBind: 寻找突变高发的一些蛋白质结合区
- SGDriver: 基于蛋白结构
- 基于DNA序列/蛋白质的边缘效应: 是否影响蛋白质与其它蛋白质的结合、转录因子与DNA的结合、miRNA与mRNA的结合
- 基于生物学通路:
- Pathscan
- CanPredict
- TransFIC
- FunSeq2
- SuSpect
Step 3: 大量突变的整体特征
- 突变特征谱(Mutation signature): C>A、C>G、C>T、T>A、T>C、T>G划分共6种不同的突变形式,加上上下游的碱基,共464=96种
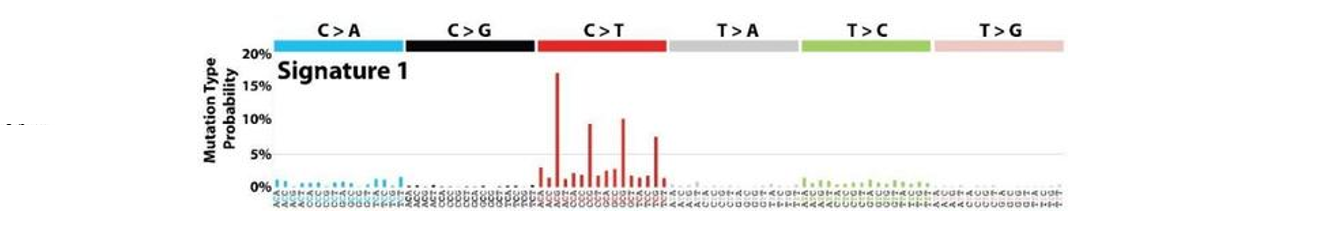
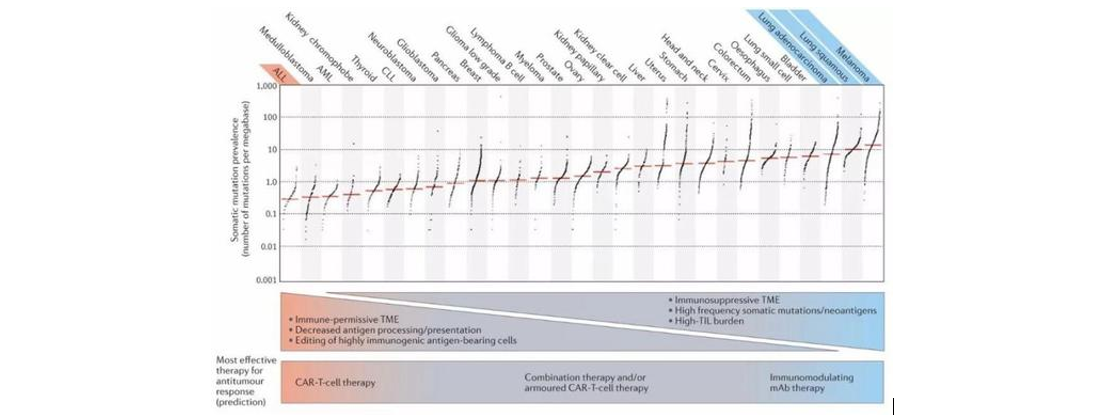
- 肿瘤突变负荷(Tumor mutation burden, TMB): 基因组上每百万碱基中,发生在体细胞编码区碱基的替换或者缺失、增加的事件数。
- 高频突变(基因)、驱动基因: 寻找突变频率显著高于背景突变频率的基因,并结合突变热点、功能、进化保守性,筛选高频突变基因,锁定driver Gene范围。软件如MutSigCV
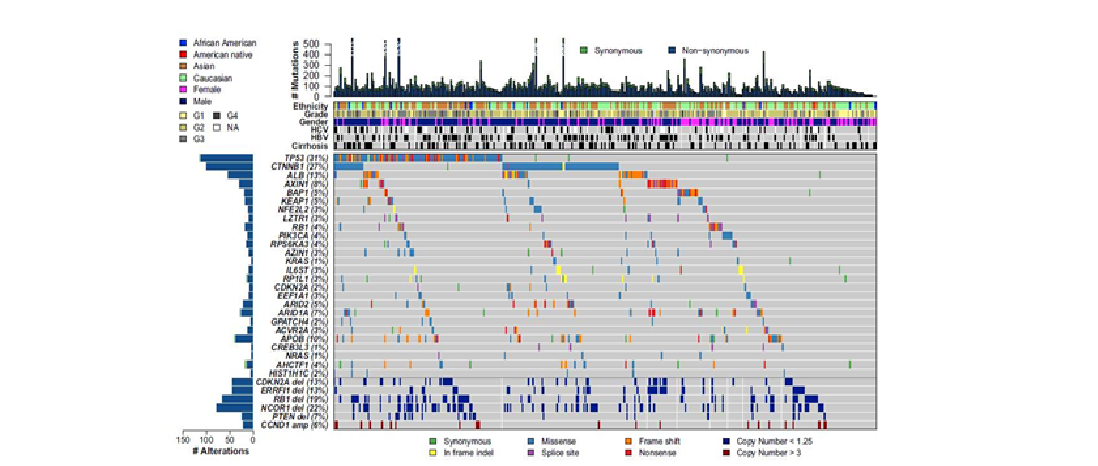
此外还有高频拷贝数变异分析、融合基因分析.

