
GATK4是最新的GATK版本,它在算法上进行了优化,运行速率得到提高,而且整合了picard。GATK4依然是用java 语言开发的,但使用方式上更加人性化,比如所有命令都是gatk cmd方式,这里的cmd是任何可以用的cmd。GATK4 的最佳实践给出了5套pipeline: Germline SNP/Indel, Somatic SNV/Indel, RNAseq SNP/Indel, Germline CNV, Somatic CNV。本文是前段时间参与Broad和Intel中国在北京的培训班上的精简记录,供自己参考用,主要是我所关注的SNV/Indel。![]()
背景知识
■ 建库library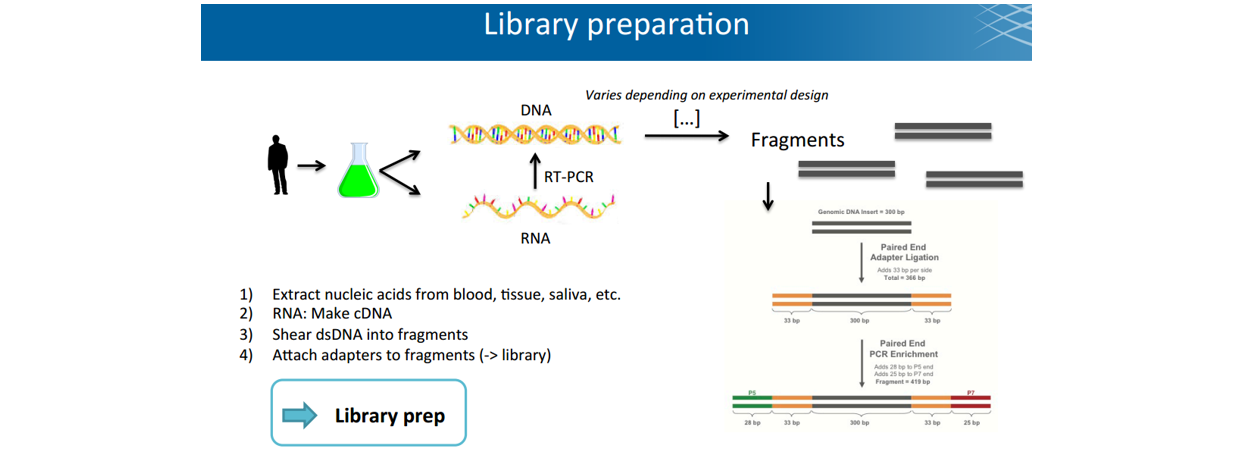
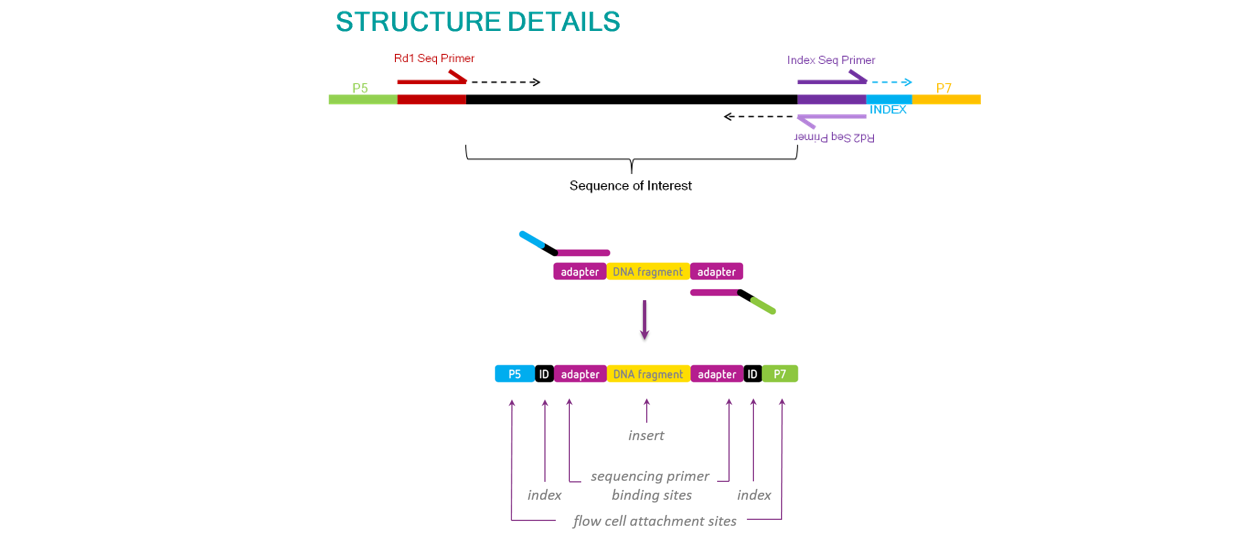
注意几个名字的理解:
- fragment修补: 末端补平、3‘加A、加接头(插有barcode)、PCR扩增(引入P5/P7)
- adaptor: 测序primer结合(Read 1 primer,Read 2 primer),其中反向adaptor也是
- index barcode:
- P5/P7: flowcell表面结合(桥式扩增时两个都用,测序时之前切断P7)
■ BAM/SAM file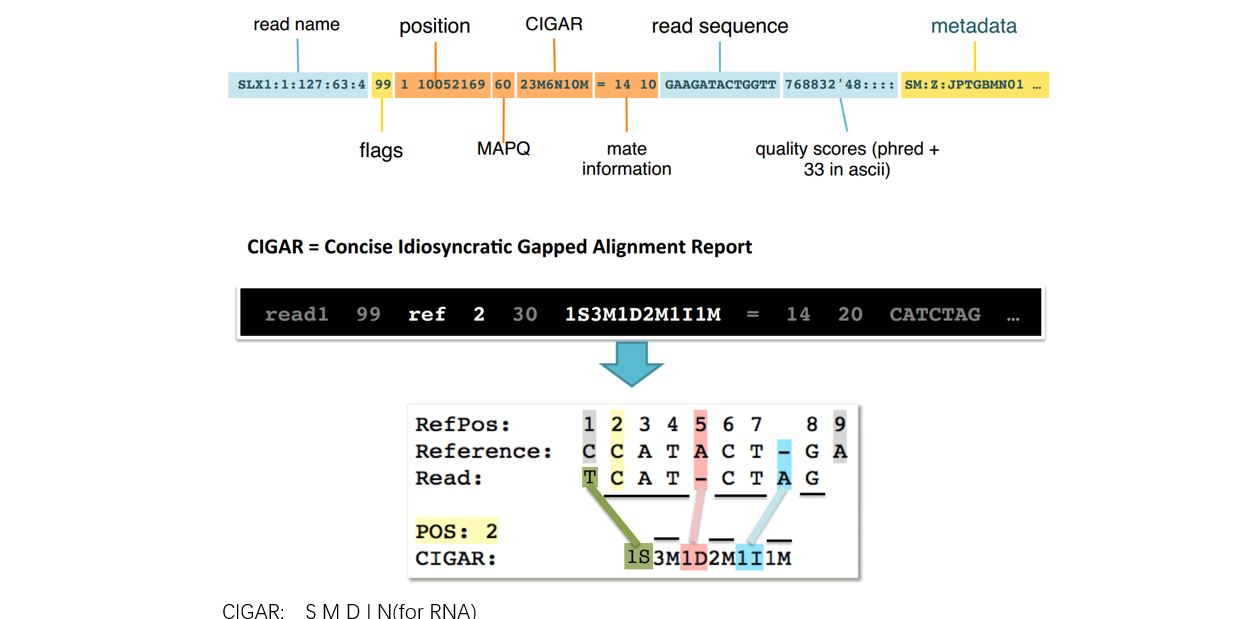
■ VCF file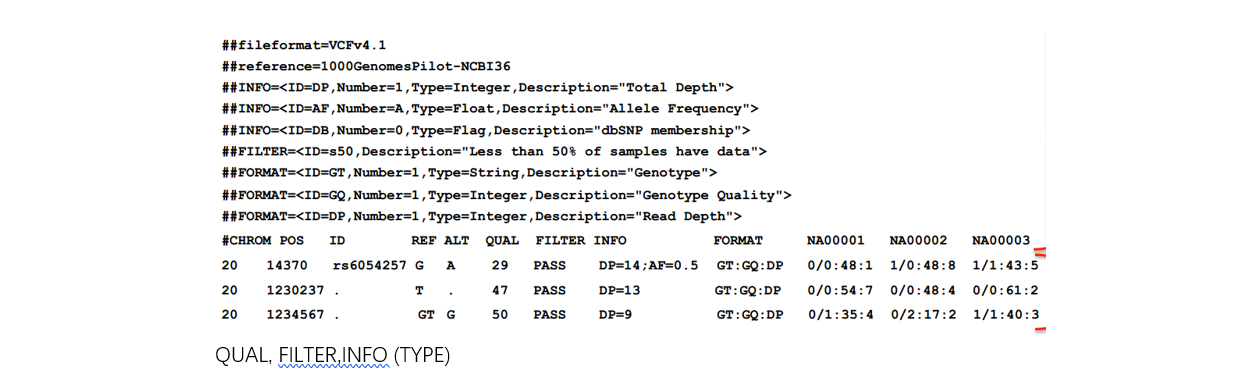
■ 不同类型的variant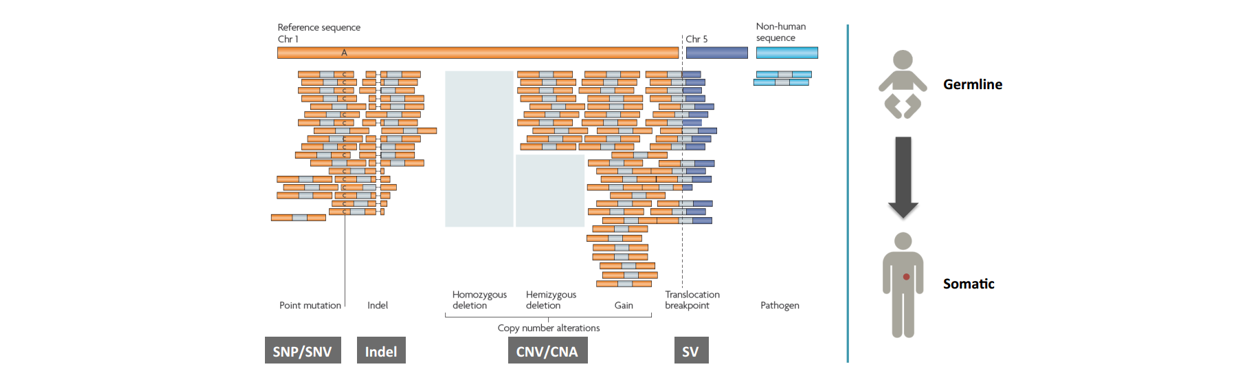
下面将要介绍的内容有:
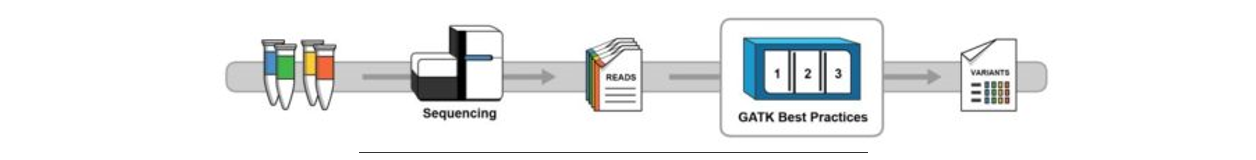
Module 1: Data Pre-processing
Prepare mapped, cleaned and sorted BAM
Step 1: MAPPING
BWA for DNA, STAR for RNA-seq
Step 2: Sort
Step 3: Mark duplicates
Step 4: BQSR
BaseRecalibrator
ApplyBQSR
Tool:AnalyzeCovariates
Notes:
alt contigs in GRCh38; the unmapped bam workflow; RNA-seq mapping
Module 2: Best Practice for Germiline SNP & InDel
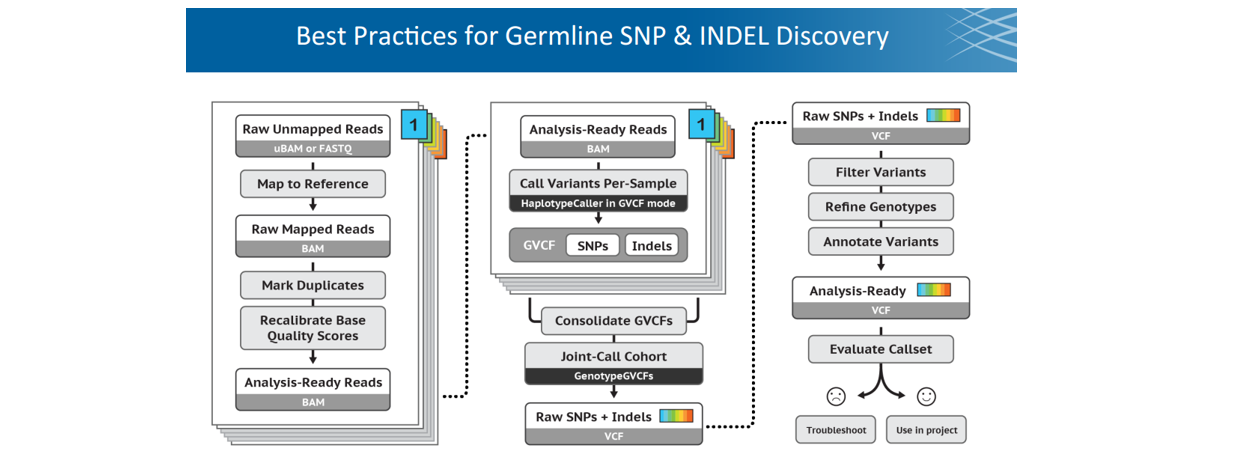
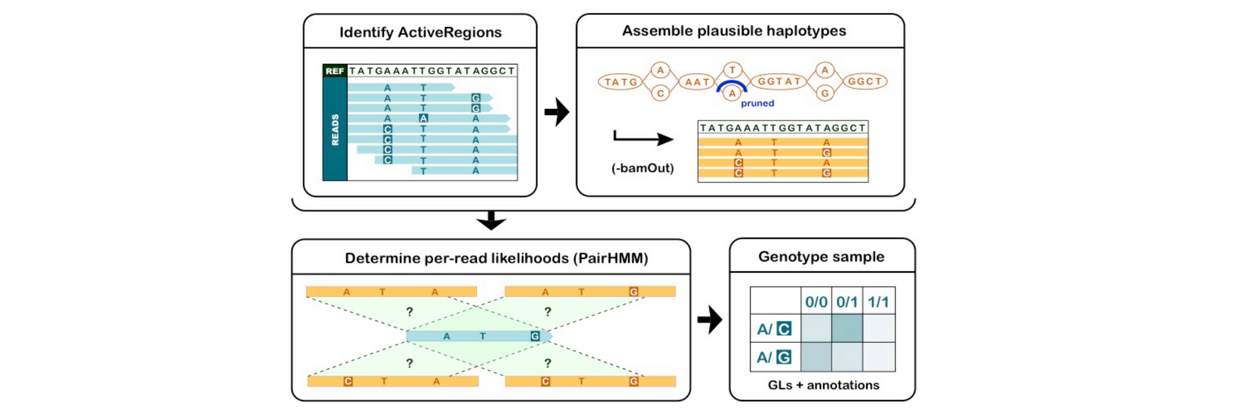
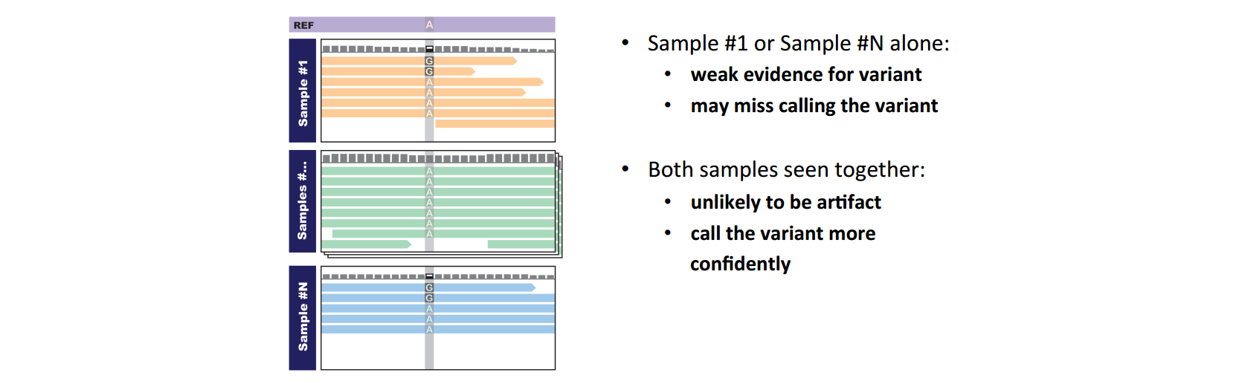
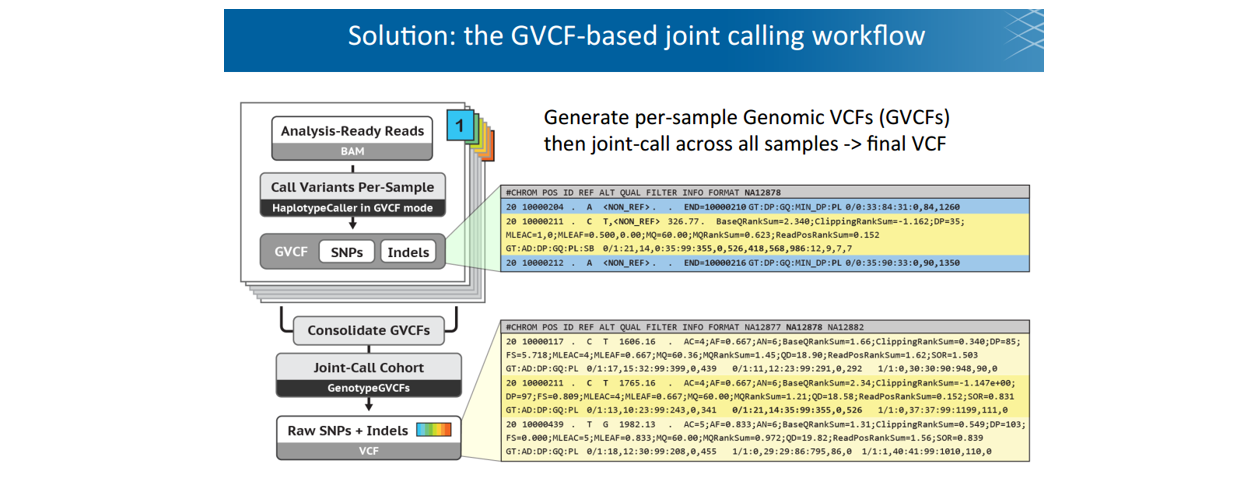
■ Step 1: Call variants with HaplotypeCaller (consolidate/joint-call with cohort)
Note: VCF和GVCF格式略不同。GVCF称为genomic VCF,里面含有Non-variant site信息,不仅仅是该样本中的SNV。相当于提前把坐标对齐了,方便后面joint-calling。包括四步操作:
- identify ActiveRegions
- assemble plausible haplotypes (local realignment, collect likely haplotypes, Smith-Waterman align)
- score haplotypes using PairHMM
- genotype each sample at each potential variant site
Default mode
12gatk HaplotypeCaller -R ref.fasta -I sample.bam -O sample.vcf-L 20:10000-11000Realignment mode and ensemble haplotypes:
12gatk HaplotypeCaller -R ref.fasta -I sample.bam -O sample.vcf#可产生多个ArtificialHaplotypeGVCF mode
123gatk HaplotypeCaller -R ref.fasta -I sample.bam -O sample.g.vcf-ERC GVCF|BP_RESOLUTION -L 20:10000-11000#GVCF for block, BR_RESOLUTION for site
GenomicsDBImport Consolidate GVCFs
GenotypeGVCFs Jointly genotype
■ Step 2: Filter (low quality)
to balance sensitivity and specifity.
two filtering approaches:
- Hard-filters using binary thresholds: Applicable to all BUT requires expertise to define appropriately
- Variant “recalibration” using machine learning: More powerful BUT requires well-curated known resources
VQSR: SNP and InDel must be separately handled

■ Step 3: Refinement
Subset callset (only SNP and only one sample)
■ Step 4: Annotate
Tabulate annotation result and Visualize
Filter variants with QUAL<30 (<10 by default)
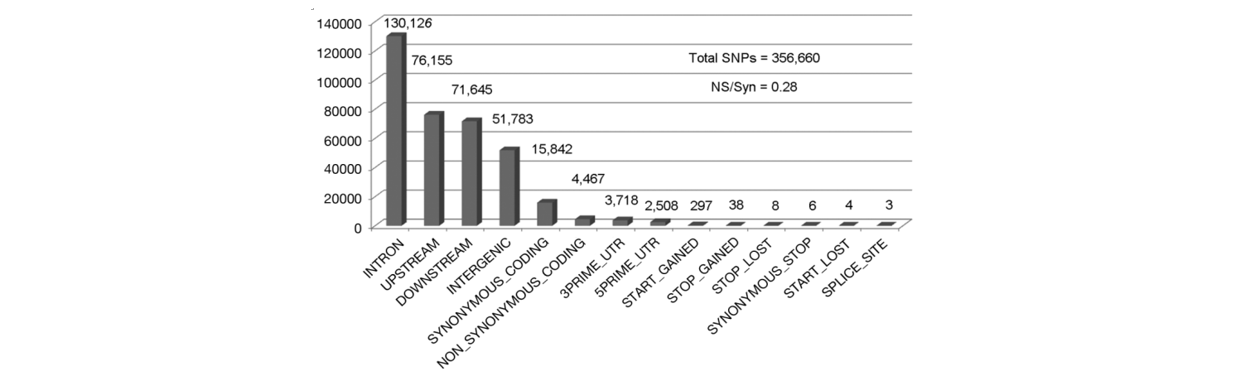
■ Step 5: Callset Evaluation
■ Step 6: check with IGV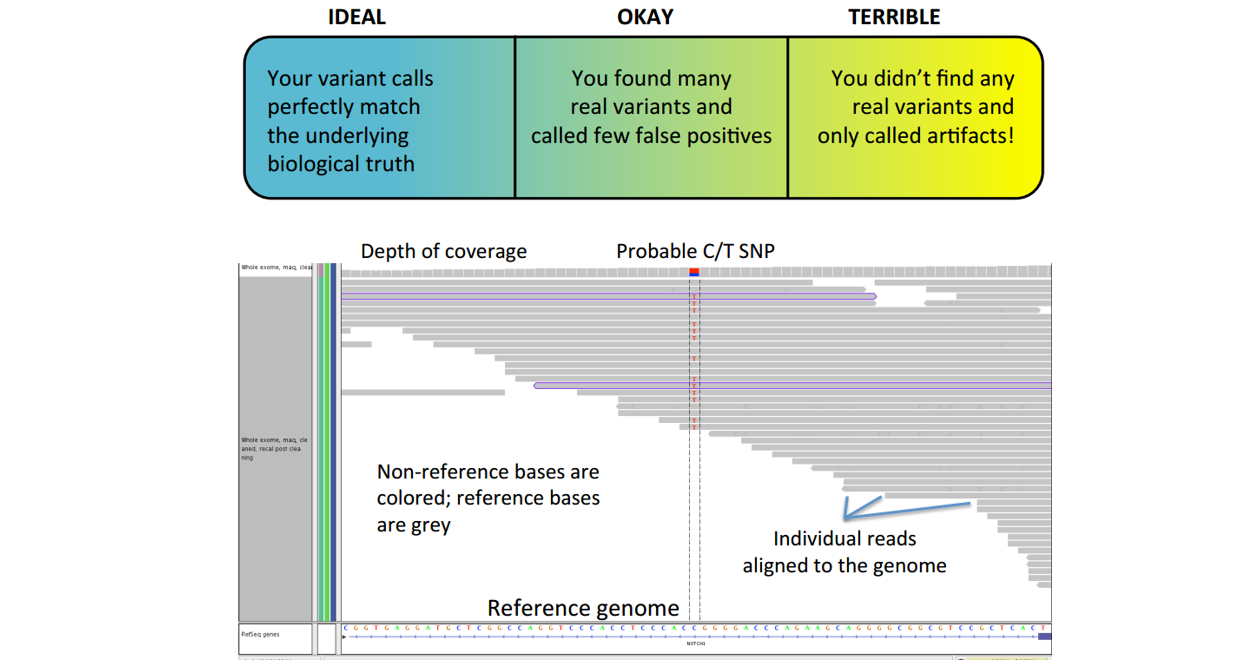
Note: Use RNA-seq data (mapping with STAR)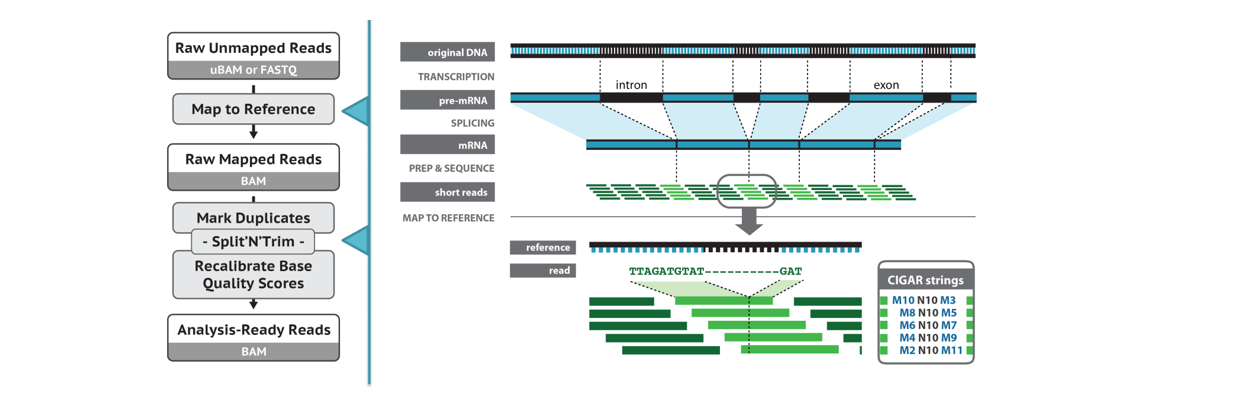
Module 3: Best Practice for Somatic SNP & InDel
different from Germline variants, due to purity (contamination of N/T) and heterogeneity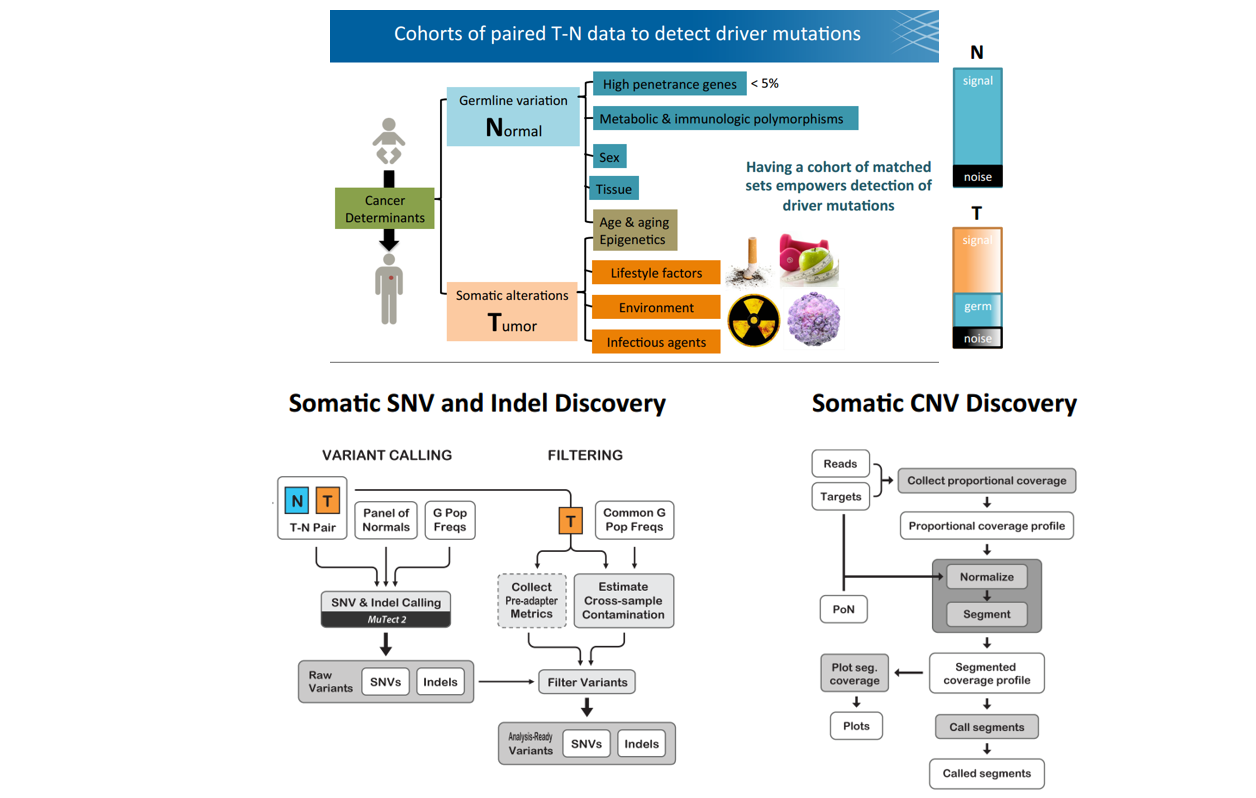
■ Step 1: Call variants with Mutect2 (denoise with normal match and PoN)
Tumor-only mode
Tumor with match mode (with realignment)
比对paired normal /a panel of normal /a germline population resource,更能发现真实的somatic SNV
■ Step 2: Filter(contamination)
■ Step3: check with IGV
Commands in GATK
Base Calling:
Copy Number Variant Discovery:
Coverage Analysis:
Diagnostics and Quality Control:
Intervals Manipulation:
Metagenomics:
Other:
Read Data Manipulation:
Reference:
Short Variant Discovery:
Structural Variant Discovery:
Variant Evaluation and Refinement:
Variant Filtering:
Variant Manipulation:

