
研究蛋白质和配体的结合模式,最重要的两个计算生物学方法是分子对接(Molecular Docking)和分子动力学模拟(Molecular Dynamic Simulation, MD)。两者各有特点,相互补充。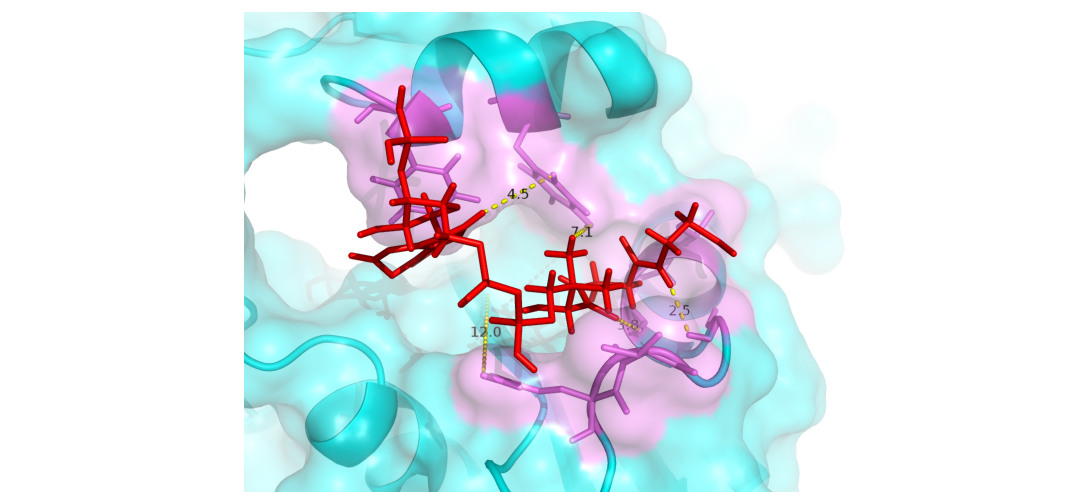
现有的解释蛋白-配体结合的理论有两种: 构象选择和诱导匹配。构象选择认为,在配体不存在时, 蛋白质处于多种构象状态的动态平衡中, 其中包括与配体结合的那个构象。相反地,诱导匹配认为,配体诱导蛋白质构象变化,从而产生与配体匹配的那种构象。诱导匹配起初用于解释酶活性的机制。不管怎样,蛋白-配体各自结构的动态变化(即下文的柔性),以及结合模式的动态变化,是研究的难点,是实验生物学无法企及的。分子对接和分子动力学模拟,可以很好解决这类问题。
最重要的因素:
构象和时间,这两个因素是在分子对接和分子动力学模拟中最重要的。所谓构象依赖性变化和时间依赖性变化。
这两个因素共同体现在结构柔性这一特点,这是蛋白-配体复合物的结构难以确定的原因。柔性也是他们发挥功能的基础条件。例如蛋白-配体模式在信号传导或调节中重要性,体现在这些功能依赖于瞬态过程。我们不可能考虑到每一个构象,所以通常的做法是,收集具有代表性的构象, 用于预测蛋白-配体的相互作用。尤其当前,我们通常下载到的蛋白质pdb文件,是蛋白质在特定实验条件下的晶体结构,不是唯一的,更需要深深把握柔性这个特点。
因此,无论分子对接还是动力学模拟,都在努力试图解决结合时的柔性问题。
各自特点:
分子对接过程较快,可以快速获得初始的结合模式。通过打分、排序和筛选不同的构象,作为起始构象。但是无法还原蛋白-配体结合的动态变化,更无法提供细节去解释结合模式。
分子动力学模拟,可以实时模拟微观的动态变化。根据晶体结构提供的构象,或者以建模方式获得的预测构象,对蛋白质进行单独的动力学模拟,以优化蛋白质结构,产生不同的初始构象。但时间尺度是严重问题。时间过少,可能捕获不到真正起作用的构象,因此也造成一些模拟无法重复。
二者互补:
分子对接和分子动力学模拟相互配合,可以为互相对方提供不同初始条件。
蛋白质、配体可能都需要能量优化。如果蛋白质结构是通过同源建模预测的,则一定需要分子动力学模拟的优化。利用统计学思路,对若干构象进行聚类,选择特征性的构象有时也是需要的。
分子对接前,可能需要动力学模拟产生更多或更优的起始蛋白质构象,并辅以其活性位点预测工具。对接之后,还需要模拟观察微观的细节,并评估结合模式。
动力学模拟之前,可能需要分子对接提供不同的初始结合模式,多种构象可以聚类分析,增加模拟的多样性,在相同的模拟时间里提高效率;当蛋白质与新的配体结合模式未知时,特别需要分子对接准备初始的构象。
深入分析:
分子对接产生若干构象,可以进行聚类,得出主要的对接模式,分析结合模拟。主要是非键作用。
分子动力学模拟最大特点是模拟,具有动态特点,分子有有柔性。更重要的用处是观察微观机制。配套的模拟条件可以根据实际情况定制。能够模拟典型生物学问题的实验条件。例如,通过控制诸如温度、压力、原子数、离子浓度和溶剂的类型等不同因素进行实验。可以控制蛋白质的柔性程度,也可以进行拉伸动力学模拟。
为进一步确认蛋白-配体的相互作用,采用MM/PBSA等方法计算结合自由能,并进行能量分解。如得出,范德华力贡献和非极性溶剂化能是稳定复合物的主要驱动力,或某些残基在蛋白-配体结合中起到关键作用。

