
过去两个月仔细看完了两本GWAS的英文书,顺便恶补了遗传研究的知识,特别是GWAS数据处理方面和基因组注释。然后把积攒下的各种疑问跟张老师聊了聊,收获颇丰。张老师不是医学出身,但博后专门做生物信息和遗传学研究,知道这里面的各种tips和常识。这些零碎的东西和技巧性的东西不是依靠自学就能知道的,需要多年的实际经验。这也是我近几年慢慢体会到的观点,博士和博后培训始终需要指导的;仅靠自学,好多坎过不了。也知道了我所学的医学遗传学是群体遗传学,只是遗传学的一个分支,这对我自己关心的基因-环境交互作用主题打开另一扇学习的大门。正好一周前,张老师有批Affy SNP6.0芯片的数据,让我给处理一下。处理过程中,有问题直接问他。处理结果也跟他核对一下。算完后,整个过程和结果没有什么问题。
群体遗传学,包括各种研究设计和统计推断,基本是遗传流行病学的内容。胡永华老师写的中文书《遗传流行病学》里有很系统很详细的描述。但这本书是传统的流行病学书,不是”大数据”时代下的场景。当前组学条件下,科研过程中有一些重要步骤,如原始数据的预处理、GWAS”大”数据的统计处理、显著SNP位点的注释等等,胡老师的书都没有涉及,这正是生物信息学的内容。
拿到SNP芯片数据后,有几个重要的步骤,它们需要专门的软件完成: 用shapeit进行shaping、用impute2进行imputation、用plink或R语言进行statistical analysis、再用各种数据库和可视化软件进行annotation并visualization。其中的各种后勤工作,如格式转化、部分SNP的提取等可以用fcgene。统计分析时,plink可以进行各种预备性工作,而利用R的灵活性进行统计分析。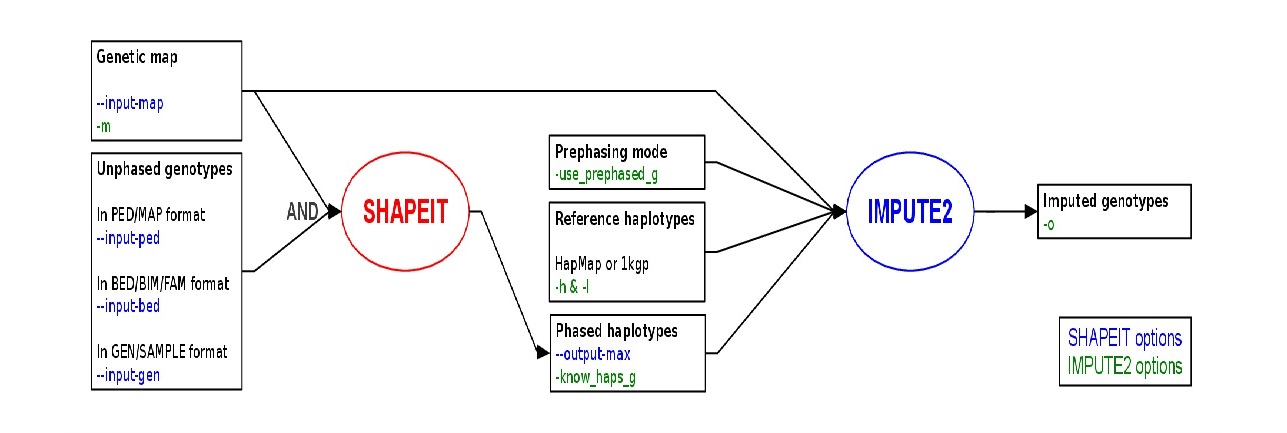
Step 1: Alignment of the SNPs: flip to fwd strand
shapeit -check -B -M -R --output-log #--input-ref
Step 2: Phasing sample [–chrX]
shapeit -B chr1_sample.bed chr1_sample.bim chr1_sample.fam \ --impute-ped --impute-vcf
-M chr1_reference.map
-O chr1_sample_phased
--thread 8
shapeit -B gwas \
-M genetic_map.txt \
-R --input-ref reference.haplotypes.gz reference.legend.gz reference.sample \
-O gwas.phased.with.ref
Step 3: Imputation
impute2 -use_prephased_g [--chrX]
-known_haps_g gwas.phased.haps
-h reference.hap
-l reference.leg
-m genetic_map
-int 9.1e6 9.6e6
-Ne 20000
-o gwas.imputed
Step 4: plink
详细的步骤可以看它们的帮助文件。通过程序的名字理解数据处理的过程是个很好的选择。图1的Manhattan图往往是GWAS统计分析的第一个结果。

